
Executive Board Advisor: Nishi Jain
Co-Authors: Vivek Babu, Collins Kariuki, Carolina Guerrero, Alex MckOsker, Daniela Galvez-Cepeda, Anahita Kodali, Sonia Fung, Maeen Arslan

Figure 1:PCR is a widely used technique that was developed in the 1970s to replicate DNA for experimental purposes
Source: Pixabay
DNA has long been an enigma, evading the understanding of scientists for decades after it was first uncovered in the 19th century. When its structure was finally resolved with contributions from Rosalind Franklin, James Watson, and Francis Crick in the mid-20th century, the scientific world was able to put an image to the word. At the very basic level, DNA is comprised of molecules called nucleotides, which contain a phosphate group, a sugar group, and a characteristic nitrogen base. Within DNA there are four kinds of nitrogen bases: adenine, guanine, thymine, and cytosine. DNA is double-stranded, meaning that the molecule consists of two nucleotide strings (bound by phosphodiester bonds). Each strand is comprised of a series of the four characteristic bases, with the two strands connected by hydrogen bonds between either adenine and thymine (which exclusively bind to one another) or cytosine and thymine (which also exclusively bind to one another), meaning that they are complementary. The ordering of these four bases within the two strands comprises the genetic code of an individual. The two strands are also arranged as a spiral—and it is for this reason that DNA is referred to as the “double helix.” However, if DNA strands really were present in real life as commonly depicted in diagrams, they would be far too large to fit inside human cells. In reality, the DNA double helix is coiled tightly to fit into the chromosomal shape that is then found in nuclei.
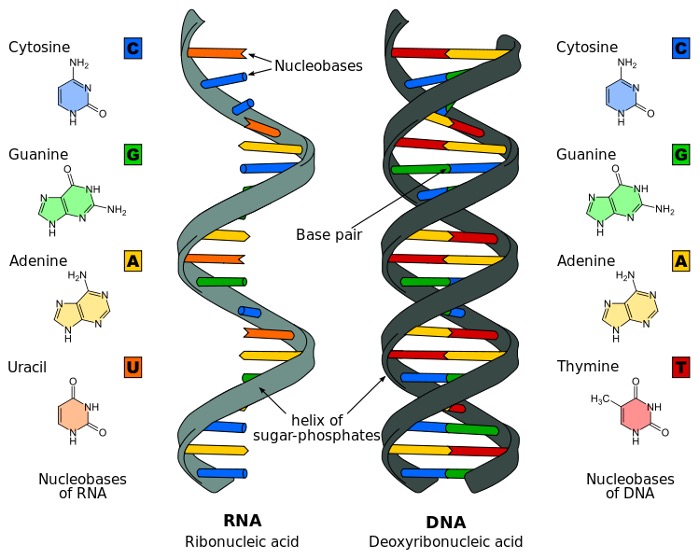
Figure 2: Structure of DNA
Source: Wikimedia Commons
Polymerase Chain Reaction (PCR) is a simple enzymatic assay which allows for a rapid amplification of a specific fragment of DNA. Billions of copies of this fragment are created, permitting a multitude of tests to be performed on the specific sequence. PCR involves three quick steps: denaturation, which allows DNA strands to separate; hybridization, where fragments (called primers) complementary to the target DNA bind the sequence of interest; and extension, in which the complementary fragment extends all the way down the desired DNA strand, developing a copy (Garibyan & Avashia, 2013). With every repetition of these steps, the total number of DNA molecules in the sample doubles.
PCR is widely used for DNA analysis due to its quick and easy procedure. It is very popular to diagnose diseases, clone and sequence genes, and even identify perpetrators of a crime based on DNA evidence left at the scene (Garibyan & Avashia, 2013). More specifically, in research, PCR is also utilized in various procedures to manipulate and study DNA. Three such procedures are genotyping, gene expression, and mutagenesis. Genotyping involves the use of PCR to detect sequence variations in specific cells and organisms by finding different alleles, or alternative forms of the gene between individuals. Similarly, PCR is applied to contrast different gene expressions among similar cell types or tissues at a given time. Lastly, in mutagenesis, PCR offers the possibility not only to clone a target DNA piece, but also to add desired mutations to the strand, creating a completely different gene (PCR Applications, n.d.).
Most recently, PCR use has been focused on testing for the presence of SARS-CoV-2, the virus that causes COVID-19, with a test called COVID-19 RT-PCR (Figure 2). This assay is a real-time reverse transcription PCR test for the detection of nucleic acid from SARS-CoV-2 in upper and lower respiratory specimens that is used to test either a single individual or a group of individuals in a pooled sample (“LabCorp Covid-19,” n.d.). The latter uses a matrix pooling strategy which, while it tests more specimens in less time, lacks precision. However, when combined with clinical observations, patient history, and epidemiological information, researchers express confidence that PCR tests do give trustworthy results (“LabCorp Covid-19,” n.d.).
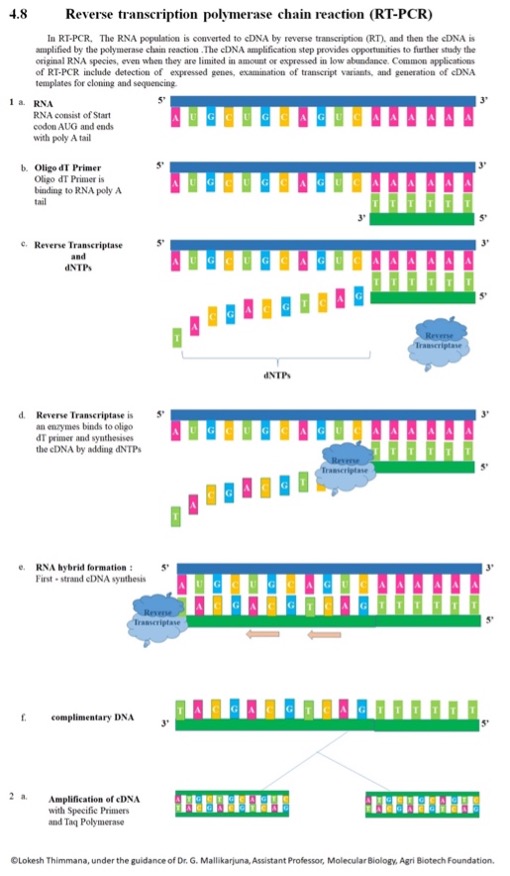
Figure 3:An Overview of RT-PCR
Source: Wikimedia Commons
As PCR becomes an increasingly popular tool for executing efficient and sensitive genomic studies, researchers have developed a large number of PCR variants. The purpose of standard PCR is to amplify and detect certain DNA and RNA sequences. The major benefit of standard PCR is that only a very small volume of genetic material is required to produce abundant and accurate PCR products. Researchers have found that more than 10 million copies of RNA or DNA molecules can be synthesized from a few copies of target sequences after just 32 cycles of amplification (Peltz, 2014). As the technology underlying standard PCR becomes more sensitive, PCR assays have made copying genetic material more cost-effective and accessible compared to other assays (Neidler, n.d.). Despite these benefits, one major limitation of PCR is the risk of contamination. Contamination from an unsterilized environment, microorganisms on operators, and cross-contamination of multiple samples can result in the wrong template being amplified, leading to inaccurate results (“Preventing Contamination in PCR Reactions”, n.d.). Another disadvantage of standard PCR is the formation of secondary structures. Because guanine/cytosine-rich (GC-rich) sequences are quite stable, they are difficult to fully separate during the denaturation phase of a PCR assay, resulting in problematic structures called hairpin loops (Neidler, n.d.). These hairpin loops block DNA polymerase (enzyme that adds DNA nucleotides) from properly synthesizing the duplicated strand. However, researchers have found that the addition of other chemicals, such as dimethylsulfoxide (DMSO), glycerol, and betaine, can disrupt these secondary structures, yielding a better outcome. (Neidler, n.d.).
Another major limitation of standard PCR is the risk of nonspecific amplification. In conventional PCR, the reaction mix is kept at room temperature; at temperatures far below the standard annealing temperature, primers tend to either anneal to DNA non-specifically or bind to one another instead of the DNA template, forming primer-dimers (“How is Hot-Start Technology Beneficial For Your PCR?”, n.d.). Because DNA polymerase will extend any segment of DNA attached to a primer, standard PCR generates undesired products that lower the true yield. To prevent nonspecific amplification, researchers have developed Hot-Start PCR. Hot-Start PCR utilizes DNA polymerase inhibitors that dissociate from DNA polymerase only at very high temperatures (Neidler, n.d.). Polymerase activity can be inhibited through several other mechanisms too including antibody interaction, chemical modification, and aptamer technology (“Hot Start PCR”, n.d.). Inhibiting DNA polymerase at room temperature allows researchers to store genetic material safely without fear that the genetic sample will continue to amplify.
History of PCR
The history of PCR dates to 1958 when chemist Kary B. Mullis invented the PCR technique while working for a biotechnology company in Emeryville, California called the Cetus Corporation (Smithsonian Institution Archives, n.d.). While the ability to produce millions of copies from meager DNA samples was revolutionary, the unfortunate reality was that Mullis’ PCR technique was very time-consuming, requiring the PCR reaction to be reset up to 40 times within four or five hours (ThermoFisher Scientific…). A common problem is the need to add a fresh enzyme at the onset of each cycle; to fix this problem Cetus engineers developed the first thermocycling machine, “Mr. Cycle,” which helped make the PCR process automated and, thus, require less labor-intensive work on the part of scientists (Smithsonian Institution Archives…).
Matters were only made better with the isolation of Taq DNA polymerase from thermophilic bacterium Thermus Aquaticus and its commercialization for popular use in 1988 by Mullis and Cetus Corporation (ThermoFisher Scientific…). The isolation of Taq polymerase was so game-changing because typical enzymes would not be able to withstand the temperature fluctuations of PCR with its heating and cooling processes as they would denature, which is why fresh enzyme had to be added for each cycle. Because the Taq polymerase is taken from a bacterium able to survive in an extreme environment like hot springs, it can withstand the fluctuating temperatures of PCR (Powledge, 2004). As such, automated PCR machines became even faster by cycling between different temperatures without enzyme replacement since the Taq enzyme could handle it. These new rapid automated PCR machines that could cycle quickly amid different temperatures were called DNA Thermal Cyclers, which were the handiwork of the Cetus Corporation and the Perkin-Elmer Corporation from Norwalk, Connecticut (Smithsonian Institution Archives…). Despite the Taq enzyme’s advantages, it wasn’t perfect. Though the Taq enzyme could withstand heat, it had low specificity and was error-prone at high temperatures. Specifically, it had a tough time amplifying guanine-cytosine base pairs at the high temperatures used.
To overcome the Taq enzyme’s reduced reliability at high temperatures, hot-start techniques were adopted in the late 1980s, where PCR reactions were heated to 95°C and allowed to cool to around 60-70°C before the addition of Taq polymerase. However, hot-start techniques ran into the same problems as early PCR in that they were time-consuming and could often cause sample cross-contamination. The next PCR innovation came in 1991 with the isolation and development of Pfu polymerases from hyperthermophilic archaeon Pyrococcus furiosus, a more thermostable enzyme compared to Taq. Pfu polymerases improved on Taq polymerases by having 3’ to 5’ exonuclease proofreading activity, which meant the ability to correct nucleotide-incorporation mistakes, a lower error rate, and greater specificity. As great as Pfu enzymes were, the use of Phusion High Fidelity DNA polymerases that was even more successful. These specially designed polymerases were made by fusing an Archae polymerase and a thermostable DNA-binding domain, which meant better proofreading ability and stability at high temperatures (ThermoFisher Scientific…). PCR continues to evolve today with the development of better polymerases and digital PCR technologies, which has been especially innovative in the analysis of single target molecules by making use of digital signals (Morley, 2014).
PCR as a Tool for COVID Testing
One critical application of PCR assays is the detection and quantification of viruses. While DNA viruses (such as herpes, smallpox, and HPV) can utilize standard PCR assays, RNA viruses (like the flu, measles, and SARS-CoV-2 virus) require the use of Reverse Transcriptase PCR (RT-PCR) (Ries, 2020). RT-PCR uses RNA (either total RNA or mRNA) to generate the template for DNA amplification. RNA is reverse transcribed into a complementary DNA – messenger RNA (cDNA-mRNA) hybrid by the enzyme reverse transcriptase (RT) which incorporates oligo(dT) primers (“Basic Principles of RT-qPCR”, n.d.). The RNAse H function of RT degrades the RNA portion of the remaining RNA segments in the sample. The single-stranded cDNA is amplified by DNA polymerase and completed to form double-stranded cDNA (Adams, 2020). While this assay allows researchers to use RNA segments, the largest constraint of RT-PCR is that RNA is difficult to work with because it is more unstable and more prone to degradation (Neidler, n.d.).
An exciting recent development in PCR technology involves assay modifications that generate more precise testing capabilities than ever seen before. One of these modifications is known as quantitative PCR (qPCR). qPCR can detect a single copy of a specific gene, and hence, is far more reliable and sensitive than conventional PCR (Augustin et al., 2017). While qPCR can be used to measure gene expression in a sample of DNA, reverse transcriptase qPCR (RT-qPCR) can use target RNA or mRNA to determine expression (Neidler, n.d.). Overall, qPCR monitors DNA or RNA amplification in real-time through fluorescence labeling. The intensity of the fluorescence signal increases proportionally to the amount of replicated DNA. In initial cycles, the signal is too low to be distinguished from the background. The point at which the signal is above the detection level corresponds to the initial number of template DNA molecules in the sample. This indicator is known as the quantification cycle (Cq) (Augustin et al., 2017). By analyzing Cq measurements, the absolute quantity or relative quantity of gene expression can be calculated.
The two standard mechanisms of qPCR are dye-based detection and probe-based assays. In dye-based qPCR, the non-specific fluorescent dyes bind throughout double-stranded DNA; once the dye binds to DNA, the fluorescence signal becomes more intense (“Real-Time qRT-PCR”, n.d.). However, a major disadvantage of dye-based detection is that only one target can be examined at a time, thus creating a more time-intensive assay. Additionally, because the fluorescence dye will bind to any DNA present within the sample, any form of contamination can severely alter the results of qPCR (Neidler, n.d.). In the second mechanism, probe-based detection, the DNA probes which consist of sequence-specific oligonucleotides are labeled with a fluorescent reporter. The intensity of the reporter only becomes detectable once a complement sequence of samples hybridizes to the sequence on the probe. Because probe-based detection specifically binds to the sample (as seen with a dye-based mechanism), probe-based qPCR is a more accurate mechanism (Neidler, n.d.). Despite this accuracy, probe-based detection is expensive because the assay requires a sequence-specific, fluorescently labeled probe, in addition to typical PCR primers (“FAQ: Should I use probe- or dye-based”, n.d.).
There are two key methods to analyze the results of qPCR: absolute and relative quantification. In absolute quantification, the Cq value of the sample of interest is interpolated with a standard curve of known samples (given by some independent means) (Augustin et al., 2017). Generally, DNA cannot be used as a standard for the absolute quantification of RNA given the variability of the reverse transcription step (“Absolute vs. Relative Quantification for qPCR”, n.d.). Despite the sensitivity of the assay and susceptibility of contamination, the precise amount of DNA copies can be found if done correctly. Additionally, relative quantification is used to compare the gene expression of two different samples. After the relative curve of the gene of interest is normalized to a housekeeping gene, the fold change (the expression ratio between the experimental control gene) can be determined. (“Absolute vs. Relative Quantification for qPCR”, n.d.).
However, there are several key limitations to qPCR. Because quantification is always analyzed on a standard curve, consistent and careful calibration is needed to properly interpolate the amount of DNA in a sample (Sedlak, 2013). Additionally, since the signal threshold is manually selected, subjectivity in PCR analysis may reduce reliability. Thus, due to these differences, inter-lab variation can be substantial, even when using commercial kits and similar protocols (Sedlak, 2013). Research has found that even within a highly trained lab with a new qPCR machine, a single assay can have 20-30% higher or lower variation in the product (Sedlak, 2013).
The limitations associated with qPCR have driven the development of digital PCR (dPCR), a novel method for precise oligonucleotide quantification. Unlike qPCR, dPCR counts the total number of individual molecules in a digital format, allowing for a more sensitive result that only requires a small amount of genetic material at the start (Prediger, 2013). dPCR functions by dividing the assay mixture into a very large number of separate small volume reactions that have either zero or one target molecule present. If the compartment contains the desired molecule, the molecule will intensely fluoresce (counted as 1 and PCR-positive); conversely, any compartment with no molecule will only display background fluorescence (counted as 0 and PCR-negative) (Prediger, 2013). Lastly, the total number of compartments that fluoresce dividing by the total measured volume results in the absolute concentration of the desired gene.
Beyond sensitivity, an added advantage of dPCR compared to standard qPCR is dPCR’s ability to multiplex without concern of cross-reactivity (Sedlak, 2013). Multiplexing is when two or more target genes are amplified in the same reaction. While multiplexing is used in qPCR, more competition for shared reagents increases the chance for non-specific interactions between primers and probes (“Principles and Uses of Multiplex PCR”, n.d.). However, in dPCR, because each reaction occurs in one compartment, different color and concentration probes can allow different target molecules to have different intensities (Prediger, 2013). While PCR’s evolution into qPCR and dPCR has allowed for more precise testing, improving the cost, accessibility, and sensitivity of PCR will further expand the assay’s applications.
PCR For Cancer Research
Another common application of PCR is its use to find mutations in heterogeneous cancer cells where DNA variants are present alongside the DNA of normal cells (Li, 2009). Since PCR doesn’t have an inherent selectivity towards DNA variants, both variant and wild-type DNA sequences are amplified with equal efficiency. Consequently, to identify and sequence the mutant DNA of tumor cells, several follow-up assays need to be set up. While PCR is reliable for screening somatic mutations, its use in sequencing unknown low abundance mutations is limited (Zuo, 2016).
Acquiring knowledge about the genetic and molecular characteristics of a patient’s tumor has become increasingly important with the rise in targeted therapies in cancer treatment (Luthra et al., 2009). Co-amplification at lower denaturation temperature-based polymerase chain reaction (COLD-PCR) is a single-step amplification method that amplifies both known and unknown low-abundance DNA variants, irrespective of mutation type and position. COLD-PCR uses a lower critical denaturing temperature (Tc) to enrich unknown mutations during amplification (Li, 2008).
Along with COLD-PCR, Emulsion PCR (EmPCR) is also used in clinical diagnosis. It is used to amplify DNA sequences in targeted next-generation sequencing (NGS)- based assays, facilitating high-throughput and economical sequencing of genomic regions of interest in clinical diagnostics (Metzker, 2010). In EmPCR, template DNA is diluted and compartmentalized in water droplets in a water-in-oil emulsion. Ideally, the dilution is to a degree where each droplet contains a single template molecule and functions as a micro-PCR reactor (Nakano, 2003). The two primary drawbacks of using standard PCR instead of EmPCR in NGS-based assays are the preferential application of short fragments and the formation of chimeric DNA molecules by recombination of homologous regions (Meyerhans, 1990). EmPCR effectively alleviates these limitations by separating the template molecules into numerous compartments, thereby preventing template competition and minimizing the chances of recombination (William, 2006).
To further enhance the diagnostic capacity of PCR, multiplex PCR has also been developed. In multiplex PCR more than one target sequence can be amplified by including more than one pair of primers in the reaction. Hence, it is used in multiple nucleic acid diagnostics, including gene deletion analysis and RNA detection. In infectious diseases, multiplex PCR is a useful method for identifying viruses, bacteria, and fungi (Elnifro, 2000).
The Technical: PCR Procedure
Evidently, PCR is a central method in a variety of scientific disciplines. But how exactly does it work? The first step in PCR is initialization, during which the reaction is heated to 94-98℃ for anywhere from 30 seconds up to a few minutes. Initialization activates the enzyme which then denatures the template DNA. However, this initial heating should not be done for more than 3 minutes to avoid destroying the enzymatic activity of the DNA (Lorenz, 2012). To avoid the loss of enzymatic activity in the polymerase, hot-start PCR can be used. Originally, Taq DNA polymerase was added into a reaction, but now more thermostable DNA polymerases are available that are even more active and stable at high temperatures, (Evans, 2009).
The next step is denaturation, which is performed at 94-98℃ and is repeated 25 to 35 times (Lorenz, 2012). This step lasts 10 seconds up to one minute, but heating should be longer if there is a high G-C percentage. Denaturation prepares the materials for annealing, which is done at 5℃ below the melting temperature (Tm) of the primers—ideally 52-58℃ (Evans, 2009). Annealing can also be done at higher temperatures to improve specificity, though this shift comes at the expense of sensitivity. In other words, sensitivity, which is finding a gene that does influence the presence of a disease, is compromised for specificity, which is finding a gene that does not influence the presence of a disease. The initial annealing temperature can be set to 5-15℃ above the Tm and is then reduced by 0.5-1.0℃ every PCR cycle until 5℃ below Tm is reached (Evans, 2009). In the early stages, this approach promotes primers annealing with high specificity to their target. In the final stages, the PCR cycle favors primer amplification which encourages higher sensitivity. Annealing is also repeated 25 to 35 times and provides a product for the remainder of the reaction.
After the primers have annealed to the template strand, the temperature of the PCR machine is increased to 72°C. At this temperature, a polymerase enzyme, usually Taq Polymerase, binds to the primers and uses the dNTPs within the reaction tube to build a strand complementary to the template strand, extending or elongating the primers from the 3’ end (“Polymerase Chain Reaction,” 2018). This process requires 1 minute to elongate the first 2 kb of DNA and an additional minute per additional 1 kb (Lorenz, 2012).
Following the use of the polymerase and the action of primers, the primary process of ‘elongation,’ or addition of nucleotides in creation of another DNA strand, ensues. This elongation process is the primary method by which DNA is replicated, as it creates the complementary strand to the template. After elongation, a complementary DNA strand exists for each template strand from the original DNA. At this point, the PCR cycle will repeat, returning to the denaturation step. After denaturation opens up each double strand of DNA into single strands, there are twice as many template strands present in the reaction; the newly synthesized DNA strands can also act as templates. As a result, the amount of DNA in the reaction effectively doubles each cycle. Generally, 25 to 30 cycles of PCR are run in order to create a large quantity of DNA while avoiding creating unwanted products (Lorenz, 2012).
On the last PCR cycle, the final elongation period is extended to 5 minutes or longer in order to ensure that all strands are completely synthesized and, when using Taq Polymerase, to add adenine residue to the 3’ end of all products, which can be useful for later procedures involving genomic or protein expression. After this is completed, the reaction is cooled to 4°C and held at this temperature for an indefinite amount of time; because PCR as an automated process is often run overnight or otherwise unsupervised, this final hold period allows the product to be stored until it is removed from the machine (Lorenz, 2012).
The Technical: PCR Reagents
There are several biological reagents that are used in PCR. The first is the DNA template strand. As discussed previously, the fundamental principle behind PCR is synthesis of DNA via the DNA polymerase enzyme. To start this synthesis, DNA polymerase needs a sample of DNA to act as a template that contains the target sequence to be amplified. At the start of the reaction, the template is subjected to high temperatures – this causes the DNA to denature, and its two strands separate to make it single-stranded (“Polymerase Chain Reaction,” n.d.).
After the template has been denatured, the mixture containing the single-stranded DNA is cooled. Primers, which are small pieces of single stranded DNA that have sequences complementary to the template strand, begin to anneal to the template (“Polymerase Chain Reaction,” n.d.). Designing the correct primer to use in the process can be quite challenging; it is arguably the most difficult part of the PCR process. One primer should anneal to one stand at the start of the DNA target sequence while the other should anneal to the other strand at the end of the target sequence. There are also several other factors that need to be taken into consideration, including: primer length, which is optimally between 15-30 nucleotide bases; G-C content, which is optimally between 40-60%; and potential for problems, including self-annealing of primers and creation of primer dimers (Lorenz, 2012).
After the template DNA has been denatured and annealed, the primers are extended to produce complementary DNA strands. Deoxyribonucleotide triphosphates, or dNTPs, are compounds that provide the actual nucleotides required for the replication process (“Polymerase Chain Reaction | PCR Process & Guide,” 2020). The effective concentration of dNTPs required for PCR depends on the length of the fragment to be amplified – a longer fragment will require a higher concentration of dNTPs to be amplified to the desired amount (“Polymerase Chain Reaction | PCR Process & Guide,” 2020).
Following the primer annealing step, DNA polymerase begins the process of synthesizing new DNA molecules. In general, DNA polymerases catalyze the formation of a phosphodiester bond, which is the chemical bond between the 3’ carbon atom of one deoxyribose group to the 5’ carbon of another. This allows the DNA strand to grow from the primer as each new dNTP is tacked on to the growing strand. In PCR, usually either one or a combination of two DNA polymerases is used. The first, Taq polymerase, is considered the standard for PCR and is the most used DNA polymerase, mainly because it allows for rapid amplification of DNA and can function in temperatures up to 97.5oC (207.5oF). The other DNA polymerase is Pfu DNA polymerase; Pfu DNA polymerase has a higher fidelity than Taq DNA polymerase because it can correct nucleotide-misincorporation issues. Pfu DNA polymerase, therefore, results in higher accuracy than Taq DNA polymerase but is significantly slower. Employing a combination of both often results in optimal synthesis, as the PCR is relatively quick and accurate (“Polymerase Chain Reaction,” n.d.; “Get To Know Your DNA Polymerases,” 2015).
There is room for error in the synthesis step, as well. DNA polymerases are known to contribute significantly to issues in the PCR process; in particular, the interaction between the DNA polymerase and the junction of the primer and template during the initiation of synthesis may be a source of amplification bias, or asymmetrical amplification of certain components of the DNA strand. Thus, this interaction must be taken into consideration when designing primers (Pan et al., 2014). After synthesis is complete, each DNA molecule is double stranded; one strand contains the old DNA, and one contains the new DNA. With each new cycle, previously synthesized DNA becomes the template, which allows for the exponential amplification of DNA that characterizes PCR.
In addition to the actual components making and being incorporated into the new DNA strand, PCR requires specific buffer solutions and ions. In chemistry, buffers are solutions that resist a change in pH; this is critical for carrying out biochemical reactions, as biological activity is often contingent on an optimal pH. For PCR, the buffer solution typically maintains a pH between 8.0 to 9.5, which is suitable for the activity of most DNA polymerases (“PCR Setup—Six Critical Components to Consider,” 2020).
Ions, which are atoms or molecules with a net electric charge, also play a crucial role in PCR. For instance, the magnesium ion (Mg2+) serves as a cofactor for DNA polymerase activity, facilitating the incorporation of dNTPs into the new DNA strand by catalyzing phosphodiester bond formation (“PCR Setup—Six Critical Components to Consider,” 2020). As a cation, or positively-charged ion, Mg2+ also stabilizes the negative charges on the phosphate backbone of the template DNA, promoting the formation of complexes between the primers and template DNA (“PCR Setup—Six Critical Components to Consider,” 2020). Some of the ions necessary for PCR may already be present in the buffer; otherwise, they must be separately added to the reaction solution (“PCR Setup—Six Critical Components to Consider,” 2020). Furthermore, the exact composition of the buffer solution and ions must be adjusted for the specific DNA polymerase being used, as different proteins work best under different conditions (“PCR Setup—Six Critical Components to Consider,” 2020).
The enhancement of PCR, especially regarding new reagents, is an active field of research. To improve PCR specificity, one research group at Xiamen University in China is studying the use of polymer-modified graphene oxide (GO) sheets (Zhong et al., 2016). The sheets, which are composed of graphene intertwined with oxygen-containing functional groups, provide an electron-rich surface that attracts positively charged DNA polymerases and magnesium ions (Zhong et al., 2016). Once the random motion of DNA polymerases and Mg2+ is reduced, favorable collisions with negatively charged reagents (e.g., DNA templates, primers, and dNTPs) may occur more frequently and with greater accuracy (Zhong et al., 2016). Favorable collisions can serve to significantly reduce amplification bias and promote faster amplification rates.
In optimizing the GO composition, the Xiamen University group experimented with different polymer modifications. One important consideration was that the GO needs to be negatively charged enough to remain water-soluble, but not so negatively charged that it repels the reagents attempting to collide with the DNA polymerases and ions held on its surface (Zhong et al., 2016). The lab found that modifying the GO with zwitterionic polymer poly(sulfobetaine), forming a material they named GO-pSB, maintained high solubility while providing a wider efficient concentration range than unmodified GO (Zhong et al., 2016). The potential impact of this research is profound, as improved specificity for PCR has significant applications in medical diagnostics and industry.

Figure 4:A standard PCR machine
Source: Wikimedia Commons
The Technical: PCR Machine
Ever since its inception in 1983, when it was first used to identify the mutation of the HBB gene that causes sickle cell anemia, the PCR machine has made considerable strides in the areas of clinical health and point-of-care applications (Zhu et al., 2020). The first PCR machine, designed by Kary Mullis and the Cetus Corporation, has undergone radical changes; current PCR machines are significantly faster and more effective than the original (Rabinow, 1996).
The Thermocycler
The term thermocycler is often used interchangeably with the terms ‘thermal cycler,’ ‘PCR machine’ or ‘DNA amplifier.’ It is used to amplify DNA segments through the Polymerase Chain Reaction. The thermocycler is the heart of the PCR process (Knoche, 2006). This is where the original DNA segments are augmented and where the reactor chips, which carry the target nucleic acid that is to be amplified, are housed. The core principle behind the operation of the thermal cycler is thermal control (i.e., temperature change), which is regulated by heating blocks (Mendoza-Gallegos et al., 2018). Because the PCR process occurs at varying temperatures, there is a need for a dependable and affordable approach to automate the temperature control function.
One method of automating temperature control involves microheater chips; thermocyclers that utilize this method are known as PCR-on-a-chip systems (Lee, 2010). Their great utility lies in their size, which has made portable PCR systems a reality. The small size of microheater chips coupled with the thin-film platinum resistors, which serve as heating elements, guarantee that the ramp rate—the maximum rate at which the thermal cycler’s heating block can change temperature—is sufficiently fast (Lee, 2010). A faster ramp rate is beneficial because it ensures a faster thermal cycling speed, an indicator of how fast DNA is being sequenced or replicated. The microheater chips offer a higher-than-normal thermal cycling speed because of the high efficacy of platinum material used.
There are a variety of characteristics of platinum that make it a good candidate for microheater chips. Platinum has a high thermal conductivity of 82.6 W/mK at a very high temperature of 1,200 K (Engineering ToolBox, 2005), which allows it to operate without thermal-induced change to chemical makeup at higher temperatures. Platinum also has a large heat of dissipation (a type of heat transfer) and a lower temperature coefficient of resistance (TCR) – the measure of the change in electrical resistance of any substance per degree of temperature change. Collectively, this means that compared to metals with a high TCR, platinum’s resistance does not increase as much with the increase in temperature so it can retain its chemical properties and not undermine the stability of the template DNA. Platinum has a lower coefficient of linear expansion too, meaning that it is not subject to a large degree of expansion over a wide range of temperatures and can maintain structural integrity (Sripumkhai, 2010).
While platinum is used for the purpose of stabilizing the PCR process and limiting exogenous effects on amplification, there are also approaches ensuring proper temperature control using a thermoelectric cooler (TEC) – normally in PCR-on-a-chip systems (Lee, 2010). For the reactions to operate correctly, the temperature needs to be adjusted at exact times and as quickly as possible. This is where TECs come in (Smith, 2013). TECs enhance temperature uniformity, which is key to temperature control. TECs do so through the Peltier effect: the cooling of one terminal and the heating of the other when an electric current is directed in a circuit of material containing two unique conductors (Lee, 2010).
Another ingenious temperature control method employs an inexpensive alternative to the commercial PCR machine. Mendoza-Gallegos and colleagues used a different-make power resistor and a computer fan as the integral parts of their off-the-shelf-made thermal cycler (Mendoza-Gallegos et al., 2018). In their design, they employed a powerful and cheap ceramic resistor to heat the target DNA solutions to high temperatures over 100 degrees Celsius and a computer fan to cool the solution in a matter of seconds. However, they noted that a bare resistor could not uniformly check the temperature distribution due to the design of the ceramic resistor. They devised an inventive plan to coat the ceramic resistor in either copper or aluminum to lessen the huge temperature difference that imperiled temperature control (Mendoza-Gallegos et al., 2018). As such, the quest for temperature control has come a long way from the very first thermocycler, which was made up of different vats of water baths; today, innovative TECs and microheater chips are used (Tan, 2017).
Optical Set-Up
Scientists who use the PCR machine need to assess the rate at which DNA amplification takes place, and they use several different fluorescence detectors to do so (Lee, 2010). Real-time thermal cyclers monitor the progress of PCR by measuring the emissions of a fluorophore—a fluorescent compound that can re-emit light upon light excitation—attached to a DNA molecule. Real-time PCR expedites the monitoring of the reaction as it progresses. One can begin with minimal amounts of nucleic acid and quantify the end product accurately (“Intro to Real-Time PCR”, n.d.).
The fluorescence emits as the PCR product accumulates with each cycle of amplification. This fluorescence is substantially enhanced when the dye is bound to double-stranded DNA (“Intro to Real-Time PCR”, n.d.). One example of fluorescence detectors are photodiodes: semiconductors which convert light energy into electrical energy (Lee, 2010). Photodiodes are preferred over common detectors such as Laser-Induced-Fluorescence (LIF) systems because the use of the latter detector is labor-intensive (Peham et. al, 2011).
Reactor Chips
Reactor chips used in PCR are commonly called Lab-on-Chip (L.O.C) devices, micro-thermocyclers, or chip thermocyclers. They are microfluidic instruments for the biochemical augmentation of DNA by thermocycling using PCR at a small scale (Li, 2008). L.O.C devices normally come in two different geometries, the first of which consists of a large cell with a thin hole, providing good temperature uniformity. The second geometry, which yields high-speed thermocycling, has a thick cell with a small diameter (“L.O.C.”, n.d.).
Efforts are being made to ensure that PCR machines are cheap, portable, and easy to maintain. For instance, Mendoza-Gallegos and colleagues have made progress not only in the miniaturization of PCR but also on reducing its cost by manufacturing various PCR parts such as the thermocycler, chassis, and optical set-up from off-the-shelf items and through 3D-printing (Mendoza-Gallegos et al., 2018). However, Mendoza-Gallegos and colleagues have made known that even though their cheaper PCR instrument is suitable enough for applications that require high initial D.N.A. quantities like pathogen detection, it is not as effective as commercial PCR instruments (Mendoza-Gallegos et al., 2018).
Limitations of PCR
Real-time PCR has proven to be an incredibly valuable technology across many fields of study. However, instrumental analysis reveals some limitations to the functionality of these methods, comparing cost, time, and figures of merit between the available technology (Powledge, 2004). Depending on available resources and funding PCR can be limited to targeting known genes, as analyzing and targeting unknown genes using PCR is often expensive and time-consuming (Smith & Osborn, 2008). PCR is also limited by the difficulty of extracting environmental samples, such as intact RNA, in order to observe patterns during cell processes (Smith & Osborn, 2008).
Real-time PCR (qPCR) is a streamlined method of DNA amplification when compared to more time and resource-consuming procedures such as competitive PCR and MPN-PCR, which require a greater amount of post-PCR analysis (Smith & Osborn, 2008). Additionally, qPCR exhibits a greater sensitivity and a wider dynamic range for immediate analysis, because the detector processes the fluorescent signal as the product forms in real-time (Smith & Osborn, 2008).
Different qPCR chemistries and techniques have unique advantages and limitations. SYBR green assays are limited by the structure of the transcript and produce a signal with a lower specificity when compared to a Taq Man assay. The Taq Man assay, however, is costly in comparison and requires more complicated temperature control throughout the procedure (Smith & Osborn, 2008). When comparing one-step qPCR to two-step qPCR, while one step is quicker, with a reduced contamination risk, the two-step qPCR is less expensive and more sensitive (Smith & Osborn, 2008). The risk of contamination is a crucial limiting factor when PCR is used for clinical diagnosis and therapy. When PCR reagents are contaminated with amplifiable DNA, the detectors can read false-positive signals that lead to misguided diagnoses (Mühl et al., 2008). Despite certain advantages, real-time PCR is limited by the accuracy of gene monitoring and transcription. In contrast, the newer digital PCR (dPCR) is more focused and selective (Bernardi et al, 2015). With this dPCR technique, the presence of a gene can be determined by the number of amplified groups post-PCR, and thus has a much lower level of detection and quantification when compared to real-time PCR (Bernardi et al, 2015).
Clearly, the clinical and scientific landscape associated with the advent, optimization, and application of PCR has seen several changes and is continuing to see several additional changes. Though the concept is deceptively simple, as its chemist and inventor of PCR Kary B. Mullis mentioned in 1958 – to throw in all ingredients needed for replication (and now plus metal ion catalysts), heat, and observe replication – modern scientists have taken the concept and applied a variety of modern technologies to ease its integration and use into several different disciplines. And although we have come a long way since 1958, we have far to go in terms of reduction of contamination, error, amplification bias, among other issues – with the pace of modern science, however, we can hold out hope that these issues may soon find resolution.
References
Adams, G. (2020). A beginner’s guide to RT-PCR, qPCR and RT-qPCR. The Biochemist, 42(3), 48–53. https://doi.org/10.1042/BIO20200034
Basic Principles of RT-qPCR – US. (n.d.). Retrieved December 31, 2020, from //www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/basic-principles-rt-qpcr.html
Berg, J. M., Tymoczko, J. L., & Stryer, L. (2002). DNA Is Replicated by Polymerases that Take Instructions from Templates. Biochemistry. 5th Edition. https://www.ncbi.nlm.nih.gov/books/NBK22513/
Bernardi, S., Di Palma, A., Tiribelli, M., Codarin, E., Ruggeri, G., Bochicchio, M. T., Malagola, M., Cattina, F., Perucca, S., Cancelli, V., Skert, C., Rosti, G., Castagnetti, F., Caimi, L., Rossi, G., Martinelli, G., & Russo, D. (2015). Digital PCR (dPCR) Overcomes the Limitations in Detection and in Quantification of Quantitative PCR (qPCR) and Reveals Different Levels of BCR-ABL1 Copies/µl Among the Chronic Myeloid Leukemia (CML) Patients Achieving Major (MR3.0) or DEEP (MR4.0, MR4.5 and MR5.0) Molecular Response with Tyrosin Kynase Inhibitors (TKIs). Blood, 126(23), 4028–4028. https://doi.org/10.1182/blood.V126.23.4028.4028
DNA polymerase preference determines PCR priming efficiency. (n.d.). Retrieved December 19, 2020, from https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3937175/
Engineering ToolBox. (2005). Thermal Conductivity of Metals, Metallic Elements and Alloys [Html]. The Engineering Toolbox. https://www.engineeringtoolbox.com/thermal-conductivity-metals-d_858.html
Erlich, H. A. (1989). Polymerase chain reaction. Journal of Clinical Immunology, 9(6), 11.
Garibyan, L., & Avashia, N. (2013). Research Techniques Made Simple: Polymerase Chain Reaction (PCR). The Journal of Investigative Dermatology, 133(3), e6. https://doi.org/10.1038/jid.2013.1
Get To Know Your DNA Polymerases. (2015). Bitesize Bio. https://bitesizebio.com/24551/get-to-know-your-dna-polymerases/
Green, S. J., Venkatramanan, R., & Naqib, A. (2015). Deconstructing the Polymerase Chain Reaction: Understanding and Correcting Bias Associated with Primer Degeneracies and Primer-Template Mismatches. PLOS ONE, 10(5), e0128122. https://doi.org/10.1371/journal.pone.0128122
Hot Start PCR | NEB. (n.d.). Retrieved December 31, 2020, from https://www.neb.com/applications/dna-amplification-pcr-and-qpcr/specialty-pcr/hot-start-pcr
How is Hot-Start Technology Beneficial For Your PCR – US. (n.d.). Retrieved December 31, 2020, from //www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/hot-start-technology-benefits-PCR.html
Introduction to Real-Time PCR Primer Design. (n.d.). Retrieved January 1, 2021, from http://www.premierbiosoft.com/tech_notes/real_time_PCR.html
Knoche, J. (2006). Thermocycler (United States Patent Patent No. USOO7030340B2). https://patentimages.storage.googleapis.com/e7/ad/bc/caa676cddce473/US7030340.pdf
Kralik, P., & Ricchi, M. (2017). A Basic Guide to Real Time PCR in Microbial Diagnostics: Definitions, Parameters, and Everything. Frontiers in Microbiology, 8. https://doi.org/10.3389/fmicb.2017.00108
LabCorp COVID-19 RT-PCR test—EUA Summary. (2020). 28.
Lee, D.-S. (2010). Real-time PCR Machine System Modeling and a Systematic Approach for the Robust Design of a Real-time PCR-on-a-Chip System. Sensors (Basel, Switzerland), 10(1), 697–718. https://doi.org/10.3390/s100100697
Li, D. (Ed.). (2008). Encyclopedia of Microfluidics and Nanofluidics. Springer US. https://doi.org/10.1007/978-0-387-48998-8
LOC PCR – Lab on Chip PCR. (n.d.). [Html]. Lab-On-Chip.Gene-Quantification.Info. Retrieved January 1, 2021, from https://www.gene-quantification.de/lab-on-chip.html
Lorenz, T. C. (2012). Polymerase Chain Reaction: Basic Protocol Plus Troubleshooting and Optimization Strategies. Journal of Visualized Experiments : JoVE, 63. https://doi.org/10.3791/3998
Mendoza-Gallegos, R. A., Rios, A., & Garcia-Cordero, J. L. (2018). An Affordable and Portable Thermocycler for Real-Time PCR Made of 3D-Printed Parts and Off-the-Shelf Electronics. Analytical Chemistry, 90(9), 5563–5568. https://doi.org/10.1021/acs.analchem.7b04843
Morley, A. A. (2014). Digital PCR: A brief history. Biomolecular Detection and Quantification, 1(1), 1–2. https://doi.org/10.1016/j.bdq.2014.06.001
Mühl, H., Kochem, A.-J., Disqué, C., & Sakka, S. G. (2010). Activity and DNA contamination of commercial polymerase chain reaction reagents for the universal 16S rDNA real-time polymerase chain reaction detection of bacterial pathogens in blood. Diagnostic Microbiology and Infectious Disease, 66(1), 41–49. https://doi.org/10.1016/j.diagmicrobio.2008.07.011
Neidler, S. (n.d.). What are the differences between PCR, RT-PCR, qPCR, and RT-qPCR? – Enzo Life Sciences. Retrieved December 19, 2020, from https://www.enzolifesciences.com/science-center/technotes/2017/march/what-are-the-differences-between-pcr-rt-pcr-qpcr-and-rt-qpcr?/
PCR Applications—Top Seven Categories—US. (n.d.). Retrieved December 30, 2020, from //www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-applications.html
PCR Setup—Six Critical Components to Consider—US. (n.d.). Retrieved December 23, 2020, from //www.thermofisher.com/us/en/home/life-science/cloning/cloning-learning-center/invitrogen-school-of-molecular-biology/pcr-education/pcr-reagents-enzymes/pcr-component-considerations.html
Peham, J. R., Vellekoop, M. J., Nöhammer, C., & Wiesinger-Mayr, H. (2011). PCR Product Detector with LED-Photodiode Fluorescence Sensing in a Nanoliter Flow-Cell for the High-Throughput Detection of Double-Stranded DNA. Procedia Engineering, 25, 936–939. https://doi.org/10.1016/j.proeng.2011.12.230
Peltz, M. (2014). 10 Ways to Minimize Contamination in a Molecular Laboratory. Luminex Corporation. https://www.luminexcorp.com/blog/10-ways-minimize-contamination-molecular-laboratory/
Polymerase Chain Reaction (PCR). (2017). https://di.uq.edu.au/community-and-alumni/sparq-ed/sparq-ed-services/polymerase-chain-reaction-pcr
Polymerase Chain Reaction (PCR). (n.d.). Retrieved December 19, 2020, from https://www.ncbi.nlm.nih.gov/probe/docs/techpcr/
Polymerase Chain Reaction | PCR Process & Guide. (n.d.). Sigma-Aldrich. Retrieved December 19, 2020, from https://www.sigmaaldrich.com/technical-documents/articles/biology/polymerase-chain-reaction.html
Prediger, E. (2013). Digital PCR (dPCR)—What is it and why use it? Integrated DNA Technologies. https://www.idtdna.com/pages/education/decoded/article/digital-pcr-(dpcr)-what-is-it-and-why-use-it-
Preventing Contamination in PCR Reactions. (n.d.). Retrieved December 31, 2020, from https://www.laboratory-equipment.com/blog/preventing-contamination-in-pcr-reactions/
Rabinow, P. (1996). Making PCR: A story of biotechnology. University of Chicago Press.
Real-Time qRT-PCR. (n.d.). Retrieved December 19, 2020, from https://www.ncbi.nlm.nih.gov/probe/docs/techqpcr/
Ries, J. (2020,). COVID-19 Will Mutate—What That Means for a Vaccine. Healthline. https://www.healthline.com/health-news/what-to-know-about-mutation-and-covid-19
Sedlak, R. H. (2013). Viral diagnostics in the era of digital polymerase chain reaction | Elsevier Enhanced Reader. https://doi.org/10.1016/j.diagmicrobio.2012.10.009
Should I use probe- or dye-based detection for my qPCR assays? | NEB. (n.d.). Retrieved January 1, 2021, from https://www.neb.com/faqs/2016/11/14/should-i-use-probe-or-dye-detection-for-my-qpcr-assays
Smith, C. (2013). Thermal Cyclers: Behind the Technology. Labcompare. http://www.labcompare.com/10-Featured-Articles/140609-Thermal-Cyclers-Behind-the-Technology/
Smith, C. J., & Osborn, A. M. (2009). Advantages and limitations of quantitative PCR (Q-PCR)-based approaches in microbial ecology: Application of Q-PCR in microbial ecology. FEMS Microbiology Ecology, 67(1), 6–20. https://doi.org/10.1111/j.1574-6941.2008.00629.x
Smithsonian Institution Archives. (n.d.). Retrieved December 19, 2020, from http://siarchives.si.edu/research/videohistory_catalog9577.html
Sripumkhai, W., Lekwichai, A., Bunjongpru, W., Porntheeraphat, S., Tunhoo, B., Ratanaudomphisut, E., Kamsri, T., Hruanun, C., Poyai, A., & Nukeaw, J. (2010). On-Chip Platinum Micro-Heater with Platinum Temperature Sensor for a Fully Integrated Disposable PCR Module. Advanced Materials Research, 93–94, 129–132. https://doi.org/10.4028/www.scientific.net/AMR.93-94.129
Tan, K. (2017). 30 Years of Thermal Cycler Innovations #1: The World’s First Thermal Cycler and How it Advanced Science. Behind the Bench. https://www.thermofisher.com/blog/behindthebench/30-years-of-thermal-cycler-innovations-1-the-worlds-first-thermal-cycler-and-how-it-advanced-science/
The History of PCR – US. (n.d.). Retrieved December 19, 2020, from www.thermofisher.com/us/en/home/brands/thermo-scientific/molecular-biology/molecular-biology-learning-center/molecular-biology-resource-library/spotlight-articles/history-pcr.html
Zhong, Y., Huang, L., Zhang, Z., Xiong, Y., Sun, L., & Weng, J. (2016). Enhancing the specificity of polymerase chain reaction by graphene oxide through surface modification: Zwitterionic polymer is superior to other polymers with different charges. International Journal of Nanomedicine, 11, 5989–6002. https://doi.org/10.2147/IJN.S120659
Zhu, H., Zhang, H., Xu, Y., Laššáková, S., Korabečná, M., & Neužil, P. (2020). PCR past, present and future. BioTechniques, 69, 317–325
Related Posts
Overview, History, Essential Concepts, and Current Topics in Transplant Immunology
First Author: Anahita Kodali1 Co-Authors: Vaishavi Agrawal2, Vivek Babu3, Sakeena...
Read More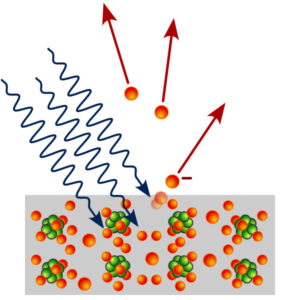
Photons Used as “Zip Ties” to Bind Electrons Together
Figure 1: The photoelectric effect as explained by Albert Einstein....
Read More
How Genetic Engineering Can Responsibly Revolutionize Healthcare Treatment
Source: iStock At just 3 months old, Victoria Gray was...
Read MoreNishi Jain, Vivek Babu, Collins Kariuki, Carolina Guerrero, Alex MckOsker, Daniela Galvez-Cepeda, Anahita Kodali, Sonia Fung, Maeen Arslan