Single Cell RNA sequencing (scRNA-Seq), a transcriptomic analysis tool, has become important in cancer research since it provides deeper insights into the genetic identity of single cells within a sample tissue. This sequencing method builds upon a widely used sequencing method—bulk RNA sequencing—that gives an averaged gene expression of each gene across all sampling cells (Shalek et al., 2014). Despite major breakthroughs in genome sequencing methods, cancer researchers have struggled to analyze the large, complex datasets emerging from scRNA-Seq of tumor samples. The key challenge is distinguishing cancer cells from nonmalignant cells since the two cell types coexist in the tumor microenvironment. To address this problem, researcher Ruli Gao and her colleagues from the University of Texas MD Anderson Cancer Center developed a specialized computational tool—CopyKAT (Copy number Karyotyping of Aneuploidy Tumors)—to find potential genetic fingerprints that differentiate cancer cells from normal cells in the tumor microenvironment.
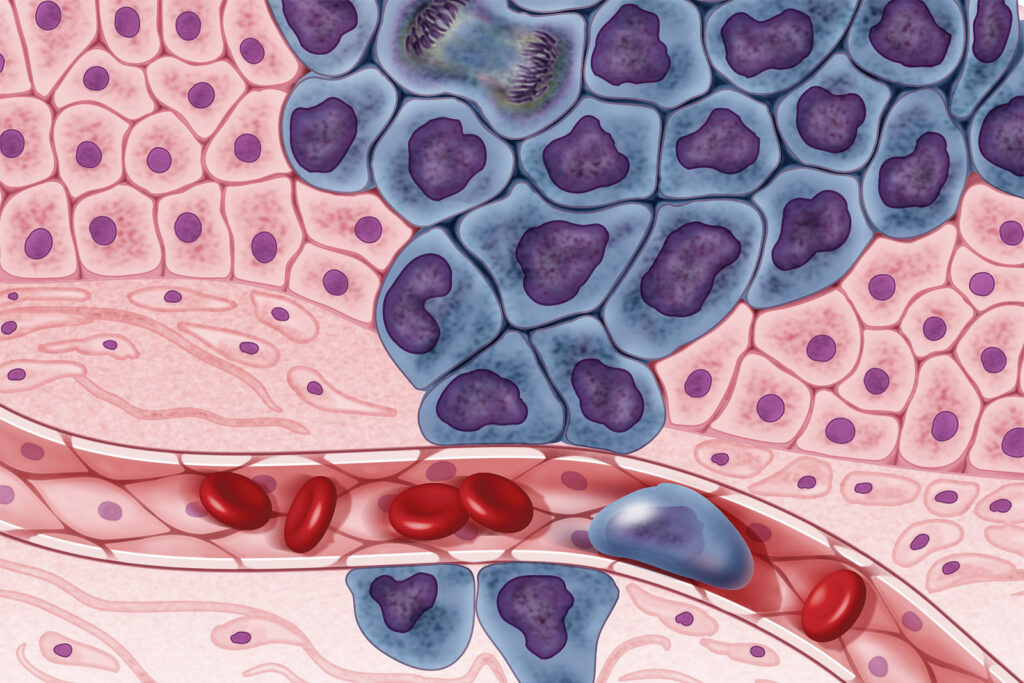
Drawing on the fact normal cells tend to be diploid (have two sets of 23 chromosomes, 46 in total) while cancer cells tend to be aneuploid (have more or less than 46 total chromosomes), which often results in uncontrollable cellular division (Figure1), CopyKAT can infer the genetic makeup of each individual cell in the tumor mass, identifying at a much higher resolution cells with an abnormal number of chromosomes (University of Texas, 2021). CopyKAT uses integrated Bayesian methods to identify genome-wide aneuploidy events with a resolution of 5Mb from scRNA-Seq data. To achieve this feat, Gao and her colleagues applied CopyKAT to infer the copy number profile of nonmalignant or diploid cells by compartmentalizing individual cells into a number of hierarchical clusters (Gao et al., 2021).
Gao and her colleagues utilized a Gaussian mixture model to evaluate the variance of each cluster. When the model is applied, the cluster with the fewest variances is selected as that containing diploid cells. However, errors may occur when the tumor sample contains a small number of normal cells, or cancer cells display very few genetic changes (Gao et al., 2021). As an alternative approach, the team evaluated the diploid status of each single cell one at a time, and a single cell with a higher diploid status was selected as a normal cell to estimate the baseline copy number values.
This tool was applied on 21 different tumors, including pancreatic cancer, triple-negative breast cancer and anaplastic thyroid cancer. CopyKAT separated cancer cells from normal cells successfully with an average of 98% accuracy for all the different datasets (Gao et al. 2021).
Another appealing feature of this computational tool is its ability to analyze the genetic diversity or heterogeneity that arises from cancer cells. Gene expression of cancer cells may differ due to cancer cells’ proliferation or external pressures. Thus, cancer cells can be organized into clonal subpopulations based on the copy number differences, which may give insights about gene expression differences between the cancer subpopulations (Gao et al., 2021). Gao and her colleagues selected and identified the clonal subpopulations by clustering single cell copy number datasets through comparing the copy number profiles of aneuploid tumor cells and diploid cells.
In summary, CopyKAT is a promising computational tool in cancer research and clinical oncology. This groundbreaking tool will greatly improve understanding of gene expression differences among tumor cells and how chromosome alterations lead to different cancer phenotypes. With information about the genetic makeup of cancer subpopulations, a few important clinical questions emerge, such as how cancerous tumors proliferate and evolve, which molecular pathways should be targeted with drugs, and how successful targeted therapy will be for a particular patient.