
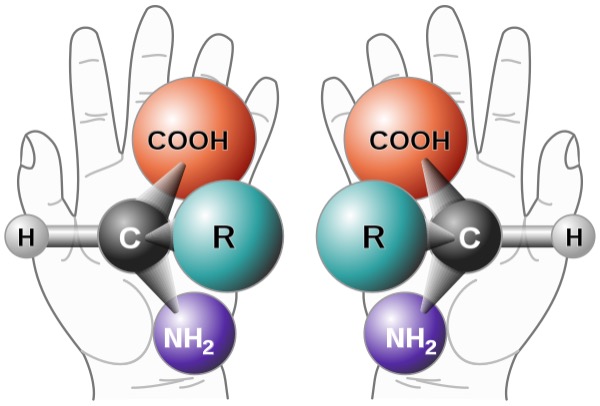
Figure 1: Two enantiomers of an amino acid. These molecules are non-superimposable mirror images of one another, in the same way human hands are. Because enantiomers can have different physiological effects, chemists have been greatly interested in designing pathways to selectively produce a desired enantiomer
Source: Wikimedia Commons
Introduction:
During the development of new synthetic reactions, chemists seek to convert simple inorganic and organic reagents into more complex products like drugs and polymers. To optimize these reactions, chemists consider the costs of input materials, time and energy requirements, and the quantity and quality of product produced (Pallardy, 2020). Many synthetic chemists are interested in changing the ratio of different chiral (or “handed” products, like human hands) enantiomeric products – molecules that are non-superimposable mirror images of each other (Hunt, 2020). In particular, the objective is enantioselective synthesis, where enantiomeric products are formed from achiral molecules – molecules that lack chiral centers like alkenes. Specifically, in enantioselective synthesis, the formation of one enantiomer is favored over another (IUPAC, 1997).
To develop enantioselective reaction schemes, scientists must consider the impact on total yield, enantiomeric excess (how much more of one enantiomer is produced over another), and the difficulty of forming and separating desired enantiomers. Scientists have several different options available for facilitating enantioselective synthesis. First, chemists can start with chiral materials – either by manipulating an already chiral starting material or adding a chiral auxiliary – a molecule that can be temporarily attached to facilitate the formation of one enantiomer (Glorius & Gnas, 2006). Second, chemists can separate mixtures of different enantiomers (racemic mixtures) via recrystallization and kinetic resolution (Robinson & Bull, 2003). Third, chemists can use chiral catalysts to facilitate the formation of a single desired enantiomeric product because of thermodynamic or kinetic favorability (Walsh, 2009). This paper will compare and contrast the benefits and drawbacks of each of these methods, as well as their ramifications for the synthesis of medicinally important compounds.
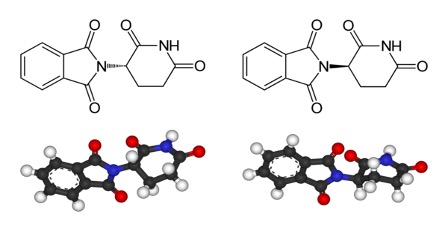
Figure 2: Enantiomers of the drug thalidomide; the left image is (S)-thalidomide and the right image is (R)-thalidomide. The primary difference between these two drugs is the 3D arrangement of their atoms; the (S) enantiomer has a “dashed” bond (indicating that the left side of the molecule is pointing “into the page”) whereas the R enantiomer has a “wedged” bond (indicating that the left side of the molecule is pointing “out of the page”). This difference in structure is responsible for the pharmacological differences of these compounds – the R enantiomer is a sedative, whereas the S enantiomer can cause birth defects
Source: Wikimedia Commons
The Importance of Enantiomers
In order to understand the importance of enantioselective synthesis, one must consider the potentially significant differences in properties when different enantiomers of the same molecule interact with a chiral biomolecule. Although their chemical formulas are identical, enantiomers differ in the arrangement of their atoms in 3-dimensional space. Additionally, though enantiomers generally have similar appearances and physical properties (besides optical rotation), they interact with other chiral molecules in different ways. For example, while the two enantiomers of the molecule carvone are both colorless oils, one smells like mint, and the other is used to flavor rye bread (Smith, 1998). As olfactory receptors are composed of chiral amino acids strung together into a chiral protein, the differing structures of the enantiomers of carvone impact interactions with these receptors, leading to differences in their smell (Feng and Zhou, 2009).
Differences in receptor interactions between enantiomer pairs not only affect their smells – they also have serious consequences for their pharmaceutical properties. One of the most infamous examples of the different biological properties of an enantiomer pair is that of Thalidomide, a drug that was prescribed as a sedative and treatment against morning sickness in pregnant women during the 1950s. However, its use was soon discontinued after over 10,000 children were born with birth defects (Franks et al., 2004). It was later determined that while the “R” enantiomer of Thalidomide acted as a sedative, the “S” enantiomer caused births defects by inhibiting proteins that facilitate the development of new blood vessels (Franks et al., 2004). Similar to Thalidomide, the biological effects of the drug Penicillamine vary greatly depending on the particular enantiomer administered. While the “D” enantiomer of the drug is effective in treating rheumatoid arthritis, the “L” enantiomer becomes toxic in the body because it inhibits the action of pyridoxine, otherwise known as Vitamin B6 (Jaffe et al., 1964). Due to the drastically different biological properties of the enantiomers of certain drugs and other chemical compounds, it is important to understand and optimize methods that can selectively produce more of a desired enantiomer.
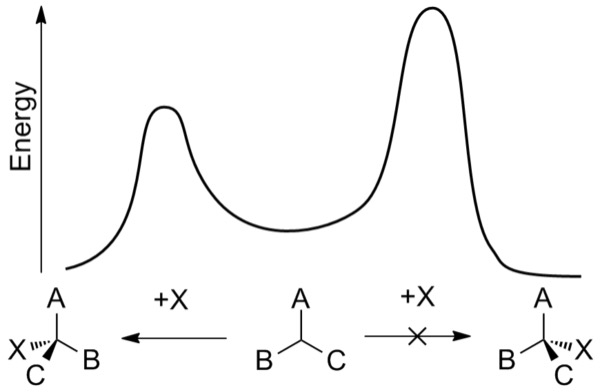
Figure 3: Energy plot of an enantioselective addition reaction. As one of the pathways (the pathway on the right) requires more energy for the reaction to occur, the pathway on the left, forming the other enantiomer is kinetically favored
Source: Wikimedia Commons
The Thermodynamics and Kinetics of Enantioselective Synthesis
At a fundamental level, the ability to control the enantioselectivity of a reaction is impacted by the kinetics and thermodynamics of the competing reactions that produce different enantiomers. In order for one reaction to outcompete the other, it must have a lower activation energy – the minimum amount of energy needed for the reaction to proceed (Wade, 2013). Chemical reactions that result in the preferential formation of a particular enantiomer do so through a process called asymmetric induction, which is influenced by a chiral stereocenter present in one of the reactants, catalysts, or the surrounding environment (IUPAC, 1994).
Donald Cram developed one of the first models for asymmetric induction in the early 1960s. Cram’s rule states simply that the stereochemistry of a new chiral center formed from a double bond is determined by the stereochemistry of an adjacent stereocenter (Cram & Elhafez, 1952). This rule is applicable to substrates that undergo nucleophilic additions such as carbonyls (C=O groups), where an electron rich substrate attacks an electron poor atom – such as the carbon in the carbonyl group which possesses a partial positive charge. Cram added that the nucleophile will attack the carbonyl from the least sterically hindered side of the molecule, meaning that it will approach the molecule from the side where the least bulky substituents are on the adjacent, or alpha, stereocenter (Cram & Elhafez, 1952). While Cram’s model successfully predicted the stereochemical configuration of 50 compounds with previously unknown configurations, it was far from perfect (Cram & Elhafez, 1952). Later work by Marc Cherest revealed that the Cram model had assumed that there was an eclipsing interaction between the “R” substituent of a carbonyl and the bulkiest substituent on its alpha carbon. This would have suggested that increasing the size of R should have decreased the stereoselectivity of reactions; in reality, increasing the size of R actually increased stereoselectivity (Cherest et al., 1968). This observation was attributed to the fact that the transition state for a nucleophilic attack is more favorable when gauche interactions – steric interactions between substituents on adjacent carbons – are minimized. When R is more bulky –an isopropyl group as opposed to a methyl substituent, for example – the gauche interactions between R and its closest substituent on the alpha carbon become more significant. Thus, the conformation of the molecule which pairs R with the smallest substituent on the same side of the planar carbonyl group is most favored (Cherest et al., 1968).
The presence of stereocenters at positions other than alpha has also been observed to cause asymmetric induction. For example, the presence of a beta stereocenter – one that is two carbons away from a carbonyl group – has been found to influence stereoselectivity in the same way as the alpha stereocenter (Evans et al., 1996). Additionally, studies conducted by Kendall Houk suggest that alkenes with chiral carbon substituents also allow for stereoselective control of that substituent if the alkene is a Z alkene – meaning the two largest substituents are on the same side of the double bond (Clayden et al., 2012). When this type of alkene undergoes electrophilic addition or epoxidation, the electron-poor electrophile must approach the alkene from its least hindered side. Therefore, Z isomers force the chiral stereocenter to assume a conformation where the smallest R group is eclipsing the double bond in order to minimize steric interactions with other alkene substituents (Clayden et al., 2012). Finally, macrocyclic systems – molecules with more than 8 atoms arranged in a ring – are also able to undergo asymmetric induction. Still and Galynker demonstrated that the addition of a single methyl substituent on a macrocyclic ring could manipulate the stereoselectivities of enolate alkylations, dimethylcuprate additions, and catalytic hydrogenations, with stereoselectivities above 90% in many cases – meaning that 90% of the compounds will be 1 stereoisomer, and 10% will be the other (Still & Galynker, 1981). As scientists are currently investigating macrocycles for their potential use in drug delivery, the ability to control macrocyclic stereochemistry via asymmetric induction is of high importance (Marsault & Peterson, 2011; Still & Galynker, 1981).
Although the enantioselectivity of a reaction is normally the same provided that the reactants involved are the same, outside forces can also change the enantioselectivity of a reaction. One of the most important considerations is the temperature of a reaction. As enantioselectivity is determined by the relative rates of competing reactions, and the difference in the natural logarithms of the rate constant is inversely proportional to temperature, a lower temperature generally leads to higher enantioselectivity (Gawley & Aube, 2012). However, there are some instances where an increase in temperature can facilitate high enantioselectivity and even change the favored product. For example, Matsumoto et al. (2016) demonstrated that the synthesis of pyrmidyl alkanols with a chiral initiator – a chiral reactant – lead to the formation of the S enantiomer with significant enantiomeric excess at 0˚C. However, at -44˚C, the R enantiomer was the most favored product. Similarly, Toth et al. (1993) reported that for the hydroformylation of styrene, as catalyzed by PtCl(SnCl3) complexes, enantioselectivity switched from 60.6% excess of the S product at 30˚C to 56.7% excess of the E product at 100˚C. Although the mechanism for this change in selectivity trends is unknown, it has been suggested that changes in the solvation of the reaction, or interaction of reactants with solvent, might cause changes in the reaction’s entropy that ultimately affect enantioselectivity (Gawley & Aube, 2012). Thus, when designing synthetic schemes to increase enantioselectivity, it is important to not only consider all reactants and catalysts, but also the reaction conditions, such as solvent type and temperature.
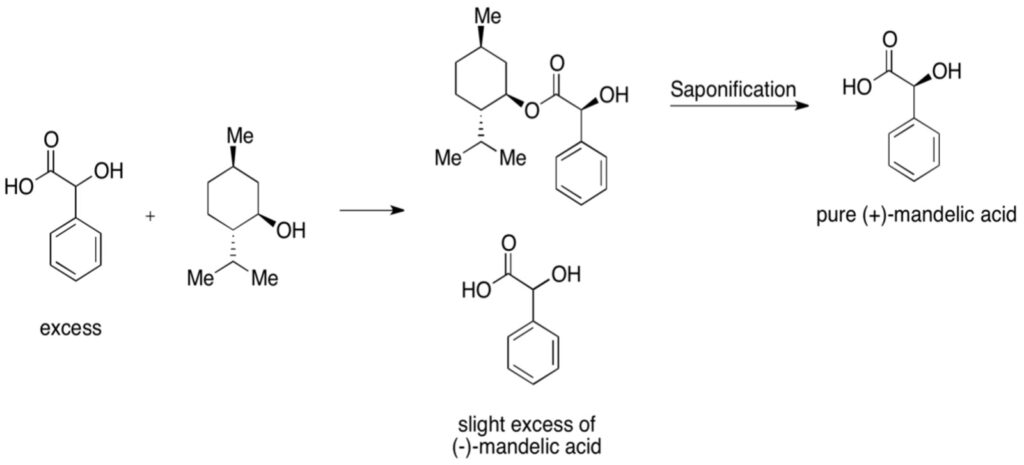
Figure 4: Example of Kinetic Resolution – mixture of mandelic acid is reacted with chiral menthol. One of the enantiomers of mandelic acid, the D enantiomer (the plus indicates the direction a solution would rotate plane-polarized light) combines more readily with pure menthol; after saponification (treatment with aqueous base), a pure mandelic acid can be produced
Source: Wikimedia Commons
Method #1: Separating Racemic Mixtures
In designing an enantioselective synthesis, there are two main ways to produce enantiomerically purified products. First, scientists can choose to develop a synthetic scheme in which chiral starting materials, catalysts, or auxiliaries are used that generate one enantiomer in excess. The second option is to separate the enantiomers of racemic mixtures – mixtures that have equal amounts of two enantiomers – from one another to make a purified final product. Both options involve the use of chiral materials – either to promote one reaction over another, or to physically separate different types of enantiomers. The methods used in separating racemic mixtures will be evaluated below.
Chiral Resolution:
Enantiomers are typically very difficult to separate from one another because many of their physical properties, like melting point and solubility, are the same (Clayden et al., 2012). In order to effectively separate a set of enantiomers, they must be converted to a diastereomer by adding a reagent that creates an additional chiral center. These diastereomers have different physical properties that allow them to be separated through conventional physical methods like crystallization and chromatography (Wyatt & Warren, 2007). The process of separating enantiomers is called chiral resolution (Clayden et al., 2012).
Effective chiral resolving agents must meet three main criteria. It should be enantiomerically pure, its reaction with both enantiomers should go to completion, and it should not become racemic under the selected reaction conditions. Additionally, the reaction to form the diastereomer should be reversible in order to produce the original enantiomer (Gawley & Aube, 2012). Many of these reagents rely upon acid-base reactions with their enantiomers. For example, in the RRR synthesis (Resolution-Racemization-Recycle synthesis) of anti-depressant duloxetine, (S)-mandelic acid reacts with a racemic mixture of the drug to produce diastereomers of the (S)-enantiomer (Fujima et al., 2006). When produced in a toluene/methanol mixture, this diastereomer is insoluble and can be filtered out; deprotonation with sodium hydroxide then removes the mandelic acid, liberating the (S)-enantiomer. This process ultimately led to a 93% purity of the (S)-enantiomer (Fujima et al., 2006).
Besides taking advantage of the differences in solubility between diastereomer products, chemists have also designed methods to separate enantiomers via chiral column chromatography. In chiral column chromatography, a racemic mixture is passed through a silica gel column with a chiral “stationary phase” of an enantiomerically pure substance like anthryl alcohol (Wyatt & Warren, 2007). One of the enantiomers will have a higher “affinity” for this stationary phase, causing it to travel down the column at a much slower speed than the other enantiomer (Clayden et al., 2012). Chiral column chromatography has proven to be an incredibly important method for separating enantiomers. For example, thanks to its resolution on a poly-N-acrylamide column, scientists found that the (+) enantiomer of the anti-malarial drug chloroquine was more effective and less toxic than the (-) enantiomer (Wyatt & Warren, 2007). Similarly, chemists at the company Dupont found that the fungicide hexaconazole could be purified to its biologically active enantiomer only through this method, as crystallization proved to be too difficult. While chiral column chromatography is efficient, it can sometimes be difficult to achieve. For example, this procedure for separating hexaconazole required precise solvent conditions, with a 918:80:2 solvent ratio of F3C-CCl3, MeCN, and Et3N, respectively (Wyatt & Warren, 2007). Additionally, it is difficult to scale chromatography separations to industrial levels, limiting its effectiveness in producing enantiomerically pure drugs (Gawley & Aube, 2012).
Kinetic Resolution:
Unlike chiral resolution, which relies upon the different physical properties of different diastereomers, kinetic resolution takes advantage of the difference in the reaction rates between different enantiomers with a common chiral catalyst or reagent. In kinetic resolution, one of the enantiomers reacts much faster than the other; over time, this results in an excess of the slower reacting enantiomer in the reaction chamber. To be practical, the starting material must be easily separated from the resolution product, and both the racemic mixture and resolving agent should be cheap (Keith et al., 2001). One of the most popular reactions in kinetic resolution is the acylation of any alcohol groups. In this method, a secondary alcohol (an alcohol with only 1 hydrogen substituent attached to the same carbon as a the -OH group) is reacted with an acid anhydride to form an ester; this process is typically facilitated by the chiral catalyst DMAP. When a chiral nucleophilic catalyst is employed, enantiomeric purity values in excess of 97% have been observed (Wyatt & Warren, 2007). This method is highly practical as the resolving agent (acetic anhydride) is fairly cheap and esters can be easily separated via fractional distillation (Keith et al., 2001). Another approach is an epoxidation reaction where a three atom cycloether is formed. This reaction has been used for the resolution of allylic alcohols (Wyatt & Warren, 2007). Chiral catalysts used for epoxidation range from titanium alkoxides to fructose-based catalysts, and allylic alcohols are generally only available in their racemic form, thus making kinetic resolution a necessity (Keith et al., 2001).
Overall, kinetic resolution is incredibly useful for enantioselective synthesis because it enables the use of cheaper and more readily available racemic mixtures of starting materials (Keith et al., 2001). However, the maximum possible yield of kinetic resolution is typically 50%, as under most circumstances the undesirable diastereomer byproduct cannot be converted back to the desired enantiomer (Keith et al., 2001).
While chiral and kinetic resolution methods are good at separating mixtures of enantiomers, they certainly have some drawbacks. Typically, the yields of both these separation techniques are 50% or less, meaning that a significant amount of starting material is wasted (Keith et al., 2001). Thus, many chemists focus on developing reactions that selectively produce one enantiomer in order to raise total yield. Special types of reagents that can produce these materials, such as chiral starting pool materials, chiral auxiliaries and catalysts, will be evaluated below.
Figure 5: The chemotherapy drug Paclitaxel can be produced from the natural terpene product verbenone, as seen on the left. As many natural products are already chiral, they can be important in facilitating enantioselective synthesis
Source: Wikimedia Commons
Method #2: Starting from Chiral Materials – The Natural Pool and Auxiliaries
When it comes to development of enantioselective schemes, many synthetic chemists have turned to using natural products and biological enzymes to facilitate enantioselective reactions. Many biological products, such as sugars and amino acids, have been used as key starting blocks (Morrow, 2016). Additionally, through improvements in biotechnology, some enantioselective methods using natural and synthetic enzymes have had great success (Jaeger & Eggert, 2004). Both of these methods will be explored further below.
Chiral Auxiliaries:
Chiral auxiliaries are enantiomers that, when added to an achiral starting material, can bias the stereochemical outcome of a reaction to form a desirable diastereomer. The auxiliary unit is then removed from the diastereomer in a way that does not cause the final product to become racemic, eliminating the need for a kinetic or chiral resolution (Gnas & Glorius, 2006). Many chiral auxiliaries are based off of simple derivatives of biological molecules, such as amino acids, carbohydrates, and terpenes, which are already chiral and abundant in nature (Diaz-Muñoz et al., 2019).
Chiral auxiliaries have had a lengthy track record of success in producing important pharmaceutical compounds. This technique was first introduced by E.J. Corey in 1975 with his enantioselective synthesis of an intermediate of prostaglandin, a hormone which can lower platelet count (Corey & Ensley, 1975). Corey’s work, which used a cyclic auxiliary called 8-phenylmenthol derived from the terpene pulegone, has proven to be very versatile in facilitating a wide range of synthetic schemes. This compound, which can facilitate stereochemical control in cycloadditions, reduction-oxidation reactions, and photochemical reactions, has enabled efficient syntheses of the glaucoma medication latanoprost and calyciphylline A, an alkaloid that has demonstrated anti-HIV activity (Diaz-Muñoz et al., 2019). Another popular class of auxiliaries are the derivatives of the heterocycle oxazolidinone, a 5 membered ring compound that has both an oxygen and a nitrogen atom in the ring. First developed by David Evans in 1981 from amino acids, this type of auxiliary has been used to facilitate asymmetric aldol reactions, alkylations, hydroxylations, and Diels-Alder cycloadditions (Heravi et al., 2016). Oxazolidinones are notable because they can facilitate the formation of two new stereocenters at the same time. As a result, this auxiliary has enabled new pathways to drugs with larger numbers of stereocenters, ranging from the anticonvulsant agent pregabalin to the antibiotic (-)-cytovaricin (Diaz-Muñoz et al., 2019).
Chiral auxiliaries are very effective in facilitating enantioselective synthesis and are reusable, unlike the reagents used in traditional resolution methods (Gawley & Aube, 2012). However, chiral auxiliaries must be used in large stoichiometric ratios to have the desired effect, and the cost of producing these auxiliaries can be quite high. Additionally, while some chiral auxiliaries can help form multiple new stereocenters, each chiral auxiliary adds at least two steps to a synthesis, which can reduce reaction yield (Gawley & Aube, 2012).
Chiral Pool Synthesis:
One of the most commonly employed methods in enantioselective synthesis is the chiral pool strategy, which draws upon the use of enantiomerically pure starting materials from nature to create enantiomerically pure compounds. Under this strategy, achiral reagents can be used to manipulate a starting material while retaining desired chiral centers (Kumar, 2014). Among the most popular starting materials for chiral pool synthesis are amino acids. Naturally used to build proteins, every amino acid except for glycine is chiral and available in an enantiomerically pure form (Clayden et al., 2012).
This strategy has been successful in developing efficient strategies for synthesizing important bioactive drugs. For instance, Aratikatla and Bhattacharya reported the synthesis of R-lacosamide, a drug used to treat epilepsy, from the amino acid serine. Unlike previous methods, which produced low yields due to the kinetic resolution of a key intermediate, the group’s synthesis produced the R enantiomer of the drug with 80% yield (Aratikatla & Bhattacharya, 2015). Similarly, Stuk et al. (1994) reported the synthesis of hydroxyethylene dipeptide isosteres, a key component of HIV protease inhibitors, from phenylalanine.
Besides essential amino acids, chemists have also turned to other functional groups from the chiral pool. Of particular interest are the terpenes, which are aromatic oils comprised of repeating C5H8 units that are commonly found in plants (Clayden et al., 2012). Syntheses performed with terpene may have incredible importance for treating both infectious diseases and cancer. For example, the anti-malarial agent (+)-cardamom peroxide was synthesized from the terpene pinene and molecular oxygen (O2) (Brill et al., 2017). Similarly, the compounds Ingenol and Englerin A, both synthesized from terpenes, have shown potential in treating renal cancer and precancerous actinic keratosis (Brill et al., 2017). On the whole, chiral pool synthesis has greatly simplified the process of manipulating and introducing chiral centers with cheap and readily available starting materials. However, it generally works best only if the starting material’s structure closely resembles the final product; otherwise, more steps are required that can drastically reduce the overall reaction yield (Morrow, 2016).

Figure 6: Example of enantioselective hydrogenation as developed by a Rhodium catalyst; the * on the product indicates the selective formation of a new stereocenter adjacent to a phenyl ring (a ring of 6 carbons bonded to 5 hydrogens). This work, as developed by William Knowles, was not initially very effective, but thanks to continued research, metal-mediated catalysts have become the method of choice for enantioselective synthesis
Source: Wikimedia Commons
Method #3: Synthetic Catalysts
Due to the low yields generated by chiral auxiliaries, modern enantioselective techniques have focused on another method: transition-metal mediated catalysis. Unlike main group elements, transition metals can easily change their oxidation states, meaning that they can accept or donate electrons as needed. Additionally, transition metals are capable of binding multiple reactants, holding them close to one another and lowering the activation energy of the reaction (Oxtoby et al., 2016). Importantly, when paired with chiral ligands – groups of atoms bound to a central atom – transition metal complexes can facilitate enantioselective reactions in a highly efficient manner. The theory, history, and method of this type of process will be explored below.
The key approach for introducing chirality into catalysts used for enantioselective synthesis is to make use of chiral C2-symmetric ligands. This means that they do not have super imposable mirror images but can be superimposed onto themselves if rotated 180˚ – the so-called C2 axis (Pfaltz & Drury, 2004). In general, transition metal complexes with these C2 symmetric ligands work by first coordinating (forming a bond) with a nucleophilic substituent on one of the reactants, like the oxygen in a carbonyl. Steric interactions between these ligands and the reactant will then typically favor the formation of one enantiomer over another due to the so-called “chiral fence,” which refers to the activation energy barriers of the different possible reaction pathways (Nishiyama et al., 1989). The C2 axis present in these molecules is crucial, as it limits the number of transition states, which can eliminate competing reaction pathways that might have lower selectivity (Pfaltz & Drury, 2004).
One particular benefit of these ligands is that some of them are considered privileged, meaning that they can be applied to the synthesis of a wide variety of enantiomeric substrates. For example, manganese complexes with the Salen ligand can catalyze the epoxidation of many alkenes with high selectivity, whereas the chromium and cobalt complexes with the same ligand can catalyze epoxide ring opening reactions (Yoon & Jacobsen, 2003). Similarly, the BINOL and BINAP ligands, which use bulky polycycles – meaning that they possess more than one connected molecular ring – are effective at catalyzing reactions ranging from hydrogenation to the Heck reaction for producing substituted alkenes (Yoon, 2003).
The development of metal-catalyzed enantioselective reactions has a long history. The first reaction was developed in 1968 by William Knowles and Leopold Horner using a chiral derivative of the commercially available rhodium catalyst [RhCl(PPh3)3] (Knowles, 2002). By switching out the three PPh3 ligands for the chiral methylpropylphenylphosphane, the group was able to hydrogenate an alkene substrate with 15% enantiomeric excess (Knowles, 2002). However, with repeated experimentation on chiral phosphorous ligands, the group was able to achieve enantioselectivities of up to 88%. Their work was eventually used by the company Monsanto in the synthesis of L-Dopa, a drug used to combat Parkinson’s Disease (Knowles, 2002). Later work from Ryoji Noyori expanded this process of hydrogenation to other substrates, such as aldehydes and ketones. Through the use of a ruthenium complex with the BINAP ligand, the group was able to effectively convert ketones and aldehydes to chiral alcohols necessary for the synthesis of the antihistamine agents orphenadrine and neobenodine (Noyori et al., 2002).
Another key advancement in the development of asymmetric catalysis occurred thanks to the efforts of Karl Barry Sharpless, who developed a series of asymmetric chiral oxidation reactions. Through the use of a titanium tartrate catalyst, Sharpless was able to transform allylic alcohols into their epoxides with over 95% enantiomeric excess (Katsuki & Sharpless, 1980). Sharpless also developed a method that could dehydroxylate, or add two alcohol groups, to an alkene using OsO4 and the readily-available quinine ligand, which has been applied in the synthesis of the anticancer agent (20S)-Camptothecin (Sharpless et al., 1994). For their combined work, Sharpless, Noyori, and Knowles were awarded shares of the 2001 Nobel Prize in Chemistry (“The Nobel Prize In Chemistry 2001”, 2001).
Due to the versatility of reactions impacted by enantioselective catalysis, many scientists have focused their efforts on expanding upon the Nobel prize-winning work. One particular point of improvement is centered on the types of metals involved in synthesis. Many of the original successful enantioselective catalysts required the use of precious and rare metals, such as osmium, rhodium and ruthenium. To make these processes more economical, scientists have focused on expanding the scope of potential metals. For example, recently scientists have designed nickel and copper-based catalysts that can selectively produce chiral phosphines like BINAP and DIPAMP, both of which are incredibly important ligands for metal-catalyzed enantioselective synthesis (Glueck, 2020). Previously, these ligands could only be produced by chiral resolutions or more expensive palladium catalysts. However, thanks to new studies in the reactivity of metal-ligand intermediates, Ni and Cu catalysts can now enantioselectively catalyze the alkylation – addition of long carbon chains – needed to form ligands like BINAP and DIPAMP (Glueck, 2020).
Studies on cheaper transition metals have also directly improved on the prior Nobel prize winning work. Previously, enantioselective hydrogenation methods relied on Rhodium based catalysts; while these were effective, Rhodium is only found in Earth’s crust at a rate of 0.0007 parts per million (Wen et al., 2021). Recent work found that by taking titanium – a metal that is more than 8 million times as abundant in Earth’s crust than Rhodium– and adding bulky substituted cyclohexanes, hydrogenation can be achieved with a 69% enantiomeric excess (Wen et al., 2021). Similarly, cobalt – a metal present in 25 parts per million – has been found to aid in the synthesis of the active agent of the anticonvulsant Levetiracetam with 98% enantiomeric excess, thanks to the introduction of bulky bisphospine ligands (Wen et al., 2021).
Conclusion:
The development of enantioselective synthesis methods has demonstrated how extensively chemical synthesis has changed over time. Initially, much of the effort in enantioselective syntheses was focused on the use of chiral auxiliaries, chiral pool synthesis, and enantiomer separation methods. However, thanks to continued innovation, scientists have been able to take advantage of chiral transition metal catalysts, which both act to limit waste and increase the versatility for synthesis plans.
References
Aratikatla, E. K., & Bhattacharya, A. K. (2015). Chiral pool approach for the synthesis of functionalized amino acids: synthesis of antiepileptic drug ( R )-lacosamide. Tetrahedron Letters, 56(42), 5802–5803. https://doi.org/10.1016/j.tetlet.2015.08.077
Chemistry (IUPAC), T. I. U. of P. and A. (1994). IUPAC – asymmetric induction (A00483). https://doi.org/10.1351/goldbook.A00483
Chérest, M., Felkin, H., & Prudent, N. (1968). Torsional strain involving partial bonds. The stereochemistry of the lithium aluminium hydride reduction of some simple open-chain ketones. Tetrahedron Letters, 9(18), 2199–2204. https://doi.org/10.1016/S0040-4039(00)89719-1
Clayden, J., Greeves, N., & Warren, S. G. (2012). Organic chemistry (2nd ed). Oxford University Press.
Corey, E. J., & Ensley, H. E. (1975). Preparation of an optically active prostaglandin intermediate via asymmetric induction. Journal of the American Chemical Society, 97(23), 6908–6909. https://doi.org/10.1021/ja00856a074
Cram, D. J., & Elhafez, F. A. A. (1952). Studies in Stereochemistry. X. The Rule of “Steric Control of Asymmetric Induction” in the Syntheses of Acyclic Systems. Journal of the American Chemical Society, 74(23), 5828–5835. https://doi.org/10.1021/ja01143a007
Diaz‐Muñoz, G., Miranda, I. L., Sartori, S. K., Rezende, D. C. de, & Diaz, M. A. N. (2019). Use of chiral auxiliaries in the asymmetric synthesis of biologically active compounds: A review. Chirality, 31(10), 776–812. https://doi.org/https://doi.org/10.1002/chir.23103
Evans, D. A., Dart, M. J., Duffy, J. L., & Yang, M. G. (1996). A Stereochemical Model for Merged 1,2- and 1,3-Asymmetric Induction in Diastereoselective Mukaiyama Aldol Addition Reactions and Related Processes. Journal of the American Chemical Society, 118(18), 4322–4343. https://doi.org/10.1021/ja953901u
Feng, G., & Zhou, W. (2019). Nostril-specific and structure-based olfactory learning of chiral discrimination in human adults. ELife, 8, e41296. https://doi.org/10.7554/eLife.41296
Franks, M. E., Macpherson, G. R., & Figg, W. D. (2004). Thalidomide. The Lancet, 363(9423), 1802–1811. https://doi.org/10.1016/S0140-6736(04)16308-3
Gawley, R. E., & Aubé, J. (2012). Principles of asymmetric synthesis (2nd ed). Elsevier.
Glueck, D. S. (2020). Catalytic Asymmetric Synthesis of P-Stereogenic Phosphines: Beyond Precious Metals. Synlett, s-0040-1707309. https://doi.org/10.1055/s-0040-1707309
Gnas, Y., & Glorius, F. (2006). Chiral Auxiliaries – Principles and Recent Applications. Synthesis, 2006(12), 1899–1930. https://doi.org/10.1055/s-2006-942399
Heravi, M. M., Zadsirjan, V., & Farajpour, B. (2016). Applications of oxazolidinones as chiral auxiliaries in the asymmetric alkylation reaction applied to total synthesis. RSC Advances, 6(36), 30498–30551. https://doi.org/10.1039/C6RA00653A
Hunt, I. (2020). Ch 7: Enantiomers. http://www.chem.ucalgary.ca/courses/350/Carey5th/Ch07/ch7-2-2.html
(IUPAC). (1997). IUPAC – stereoselective synthesis (S05990). https://doi.org/10.1351/goldbook.S05990
Jaffe, I. A., Altman, K., & Merryman, P. (1964). The Antipyridoxine Effect of Penicillamine in Man. The Journal of Clinical Investigation, 43(10), 1869–1873. https://doi.org/10.1172/JCI105060
Katsuki, T., & Sharpless, K. B. (1980). The first practical method for asymmetric epoxidation. Journal of the American Chemical Society, 102(18), 5974–5976. https://doi.org/10.1021/ja00538a077
Keith, J. M., Larrow, J. F., & Jacobsen, E. N. (2001). Practical Considerations in Kinetic Resolution Reactions. Advanced Synthesis & Catalysis, 343(1), 5–26. https://doi.org/https://doi.org/10.1002/1615-4169(20010129)343:1<5::AID-ADSC5>3.0.CO;2-I
Knowles, W. S. (2002). Asymmetric Hydrogenations (Nobel Lecture). Angewandte Chemie International Edition, 41(12), 1998–2007. https://doi.org/https://doi.org/10.1002/1521-3773(20020617)41:12<1998::AID-ANIE1998>3.0.CO;2-8
Kumar, Dr. Konda. (2014). CHIRAL SYNTHESIS: AN OVERVIEW. International Journal of Pharmaceutical Research and development. 6. 70.
Marsault, E., & Peterson, M. L. (2011). Macrocycles Are Great Cycles: Applications, Opportunities, and Challenges of Synthetic Macrocycles in Drug Discovery. Journal of Medicinal Chemistry, 54(7), 1961–2004. https://doi.org/10.1021/jm1012374
Matusmoto, A., Fujiwara, S., Hiyoshi, Y., Zawatzky, K., Makarov, A. A., Welch, C. J., & Soai, K. (2017). Unusual reversal of enantioselectivity in the asymmetric autocatalysis of pyrimidyl alkanol triggered by chiral aromatic alkanols and amines. Organic & Biomolecular Chemistry, 15(3), 555–558. https://doi.org/10.1039/C6OB02415G
Morrow, G. W. (2016). Biorganic synthesis: an introduction. Oxford University Press.
Nishiyama, Hisao., Sakaguchi, Hisao., Nakamura, Takashi., Horihata, Mihoko., Kondo, Manabu., & Itoh, Kenji. (1989). Chiral and C2-symmetrical bis(oxazolinylpyridine)rhodium(III) complexes: effective catalysts for asymmetric hydrosilylation of ketones. Organometallics, 8(3), 846–848. https://doi.org/10.1021/om00105a047
Nobel Foundation. (n.d.). The Nobel Prize in Chemistry 2001. NobelPrize.Org. Retrieved February 5, 2021, from https://www.nobelprize.org/prizes/chemistry/2001/summary/
Noyori, R. (2002). Asymmetric Catalysis: Science and Opportunities (Nobel Lecture). Angewandte Chemie International Edition, 41(12), 2008–2022. https://doi.org/https://doi.org/10.1002/1521-3773(20020617)41:12<2008::AID-ANIE2008>3.0.CO;2-4
Oxtoby, D. W., Gillis, H. P., & Campion, A. (2016). Principles of modern chemistry (Eighth edition). Cengage Learning.
Pallardy, R. (2020). Chemical synthesis. Encyclopedia Britannica. https://www.britannica.com/science/chemical-synthesis
Pfaltz, A., & Drury, W. J. (2004). Design of chiral ligands for asymmetric catalysis: From C2-symmetric P,P- and N,N-ligands to sterically and electronically nonsymmetrical P,N-ligands. Proceedings of the National Academy of Sciences, 101(16), 5723–5726. https://doi.org/10.1073/pnas.0307152101
Porter, W. H. (1991). Resolution of chiral drugs. Pure and Applied Chemistry, 63(8), 1119–1122. https://doi.org/10.1351/pac199163081119
Robinson, D. E. J. E., & Bull, S. D. (2003). Kinetic resolution strategies using non-enzymatic catalysts. Tetrahedron: Asymmetry, 14(11), 1407–1446. https://doi.org/10.1016/S0957-4166(03)00209-X
Sharpless, K. B., Kolb, H. C., & VanNieuwenhze, M. S. (1994). Catalytic Asymmetric Dihydroxylation. Chemical Reviews, 94(8), 2483–2547. https://doi.org/10.1021/cr00032a009
Shen, Z., Lv, C., & Zeng, S. (2016). Significance and challenges of stereoselectivity assessing methods in drug metabolism. Journal of Pharmaceutical Analysis, 6(1), 1–10. https://doi.org/10.1016/j.jpha.2015.12.004
Still, W. C., & Galynker, I. (1981). Chemical consequences of conformation in macrocyclic compounds: An effective approach to remote asymmetric induction11This work was presented in part at the Fourteenth Sheffield Stereochemistry Symposium in Sheffield, England, on December 17, 1980. Tetrahedron, 37(23), 3981–3996. https://doi.org/10.1016/S0040-4020(01)93273-9
Stuk, T. L., Haight, A. R., Scarpetti, D., Allen, M. S., Menzia, J. A., Robbins, T. A., Parekh, S. I., Langridge, D. C., & Tien, J.-H. J. (1994). An Efficient Stereocontrolled Strategy for the Synthesis of Hydroxyethylene Dipeptide Isosteres. The Journal of Organic Chemistry, 59(15), 4040–4041. https://doi.org/10.1021/jo00094a006
Toth, I., Guo, I., & Hanson, B. E. (1993). Influence of the reaction temperature on the enantioselection of styrene hydroformylation catalyzed by PtCl(SnCl3) complexes of p-aryl-substituted chiral ligands. Organometallics, 12(3), 848–852. https://doi.org/10.1021/om00027a038
Wade, L. G. (2013). Organic chemistry (8th ed). Pearson.
Walsh, P. J. (2009). Fundamentals of asymmetric catalysis. Sausalito, Calif. : University Science Books. http://archive.org/details/fundamentalsofas0000wals
Wen, J., Wang, F., & Zhang, X. (2021). Asymmetric hydrogenation catalyzed by first-row transition metal complexes. Chemical Society Reviews. https://doi.org/10.1039/D0CS00082E
Wyatt, P., & Warren, S. G. (2007). Organic synthesis: strategy and control. http://www.dawsonera.com/depp/reader/protected/external/AbstractView/S9780470061206
Yoon, T. P. (2003). Privileged Chiral Catalysts. Science, 299(5613), 1691–1693. https://doi.org/10.1126/science.1083622
Related Posts
Researchers produce important RNA subunit using a biocatalyst for the first time
Figure 1: The image on the left displays a standard...
Read More
Possible Biosignature for Life Discovered on Venus
Figure 1: This is an image of Venus with its...
Read More
A Spoonful of (Modified) Sugar as an Antiviral Medicine
Figure 1: A negative stain transmission electron micrograph of Influenza...
Read MoreAndrew Sasser
Comments are closed.